
Обязательная сертификация медицинских изделий
Подборка наиболее важных документов по запросу Обязательная сертификация медицинских изделий (нормативно–правовые акты, формы, статьи, консультации экспертов и многое другое).
Судебная практика
Постановление Правительства РФ от 01.06.2021 N 852(ред. от 16.02.2022)»О лицензировании медицинской деятельности (за исключением указанной деятельности, осуществляемой медицинскими организациями и другими организациями, входящими в частную систему здравоохранения, на территории инновационного центра «Сколково») и признании утратившими силу некоторых актов Правительства Российской Федерации»(вместе с «Положением о лицензировании медицинской деятельности (за исключением указанной деятельности, осуществляемой медицинскими организациями и другими организациями, входящими в частную систему здравоохранения, на территории инновационного центра «Сколково»)»)(с изм. и доп., вступ. в силу с 01.09.2022)в) наличие заключивших с соискателем лицензии трудовые договоры работников, имеющих образование, предусмотренное квалификационными требованиями к медицинским и фармацевтическим работникам, и пройденной аккредитации специалиста или сертификата специалиста по специальности, необходимой для выполнения заявленных соискателем лицензии работ (услуг);
1. Медицинскими изделиями являются любые инструменты, аппараты, приборы, оборудование, материалы и прочие изделия, применяемые в медицинских целях отдельно или в сочетании между собой, а также вместе с другими принадлежностями, необходимыми для применения указанных изделий по назначению, включая специальное программное обеспечение, и предназначенные производителем для профилактики, диагностики, лечения и медицинской реабилитации заболеваний, мониторинга состояния организма человека, проведения медицинских исследований, восстановления, замещения, изменения анатомической структуры или физиологических функций организма, предотвращения или прерывания беременности, функциональное назначение которых не реализуется путем фармакологического, иммунологического, генетического или метаболического воздействия на организм человека. Медицинские изделия могут признаваться взаимозаменяемыми, если они сравнимы по функциональному назначению, качественным и техническим характеристикам и способны заменить друг друга.
2. Медицинские изделия подразделяются на классы в зависимости от потенциального риска их применения и на виды в соответствии с номенклатурной классификацией медицинских изделий. Номенклатурная классификация медицинских изделий утверждается уполномоченным федеральным органом исполнительной власти.
(в ред. Федеральных законов от 25.11.2013 N 317-ФЗ, от 11.06.2021 N 170-ФЗ)
(см. текст в предыдущей редакции)
3.1. Допускаются предусмотренные нормативной, технической и (или) эксплуатационной документацией производителя (изготовителя) транспортировка, монтаж, наладка, настройка, калибровка медицинского изделия и иные действия, необходимые для ввода медицинского изделия в эксплуатацию, применение, эксплуатация, в том числе техническое обслуживание, и ремонт медицинского изделия по окончании срока действия регистрационного удостоверения на это медицинское изделие, если срок службы (срок годности) медицинского изделия не истек.
(часть 3.1 введена Федеральным законом от 30.04.2021 N 128-ФЗ)
3.2. До истечения срока службы (срока годности) медицинских изделий допускается обращение таких изделий, в том числе произведенных в течение ста восьмидесяти календарных дней после дня принятия уполномоченным федеральным органом исполнительной власти решения о внесении изменений в документы, содержащиеся в регистрационном досье на медицинское изделие, в соответствии с информацией, содержащейся в таких документах до дня принятия указанного решения.
(часть 3.2 введена Федеральным законом от 30.04.2021 N 128-ФЗ)
3.3. Действие требований, установленных частью 3 настоящей статьи, может быть изменено в отношении участников экспериментального правового режима в сфере цифровых инноваций в соответствии с программой экспериментального правового режима в сфере цифровых инноваций, утверждаемой в соответствии с Федеральным законом от 31 июля 2020 года N 258-ФЗ «Об экспериментальных правовых режимах в сфере цифровых инноваций в Российской Федерации», с учетом требований, установленных правом Евразийского экономического союза.
(часть 3.3 введена Федеральным законом от 02.07.2021 N 331-ФЗ)
4. На территории Российской Федерации разрешается обращение медицинских изделий, прошедших государственную регистрацию в порядке, установленном Правительством Российской Федерации, и медицинских изделий, прошедших регистрацию в соответствии с международными договорами и актами, составляющими право Евразийского экономического союза. Действие данных требований может быть изменено в отношении участников экспериментального правового режима в сфере цифровых инноваций в соответствии с программой экспериментального правового режима в сфере цифровых инноваций, утверждаемой в соответствии с Федеральным законом от 31 июля 2020 года N 258-ФЗ «Об экспериментальных правовых режимах в сфере цифровых инноваций в Российской Федерации», с учетом требований, установленных правом Евразийского экономического союза.
(в ред. Федеральных законов от 30.04.2021 N 128-ФЗ, от 02.07.2021 N 331-ФЗ)
5. На территории Российской Федерации не регистрируются:
1) медицинские изделия, перечисленные в пункте 11 статьи 4 Соглашения о единых принципах и правилах обращения медицинских изделий (изделий медицинского назначения и медицинской техники) в рамках Евразийского экономического союза от 23 декабря 2014 года;
Со 02.01.2027 п. 2 ч. 5 ст. 38 утрачивает силу (ФЗ от 30.04.2021 N 128-ФЗ).
2) медицинские изделия, ввезенные на территорию Российской Федерации для оказания медицинской помощи по жизненным показаниям конкретного пациента на основании разрешения, выданного в порядке, установленном Правительством Российской Федерации, уполномоченным федеральным органом исполнительной власти, осуществляющим функции по контролю и надзору в сфере охраны здоровья;
Со 02.01.2027 п. 3 ч. 5 ст. 38 утрачивает силу (ФЗ от 30.04.2021 N 128-ФЗ).
3) медицинские изделия, произведенные в Российской Федерации для экспорта за пределы территории Евразийского экономического союза и не предназначенные для применения на территории Евразийского экономического союза, а также произведенные в Российской Федерации для проведения опытно-конструкторских работ, исследований (испытаний);
4) медицинские изделия, которые предназначены для применения на территории международного медицинского кластера или на территориях инновационных научно-технологических центров;
Со 02.01.2027 п. 5 ч. 5 ст. 38 утрачивает силу (ФЗ от 30.04.2021 N 128-ФЗ).
5) медицинские изделия, представляющие собой укладки, наборы, комплекты и аптечки, состоящие из зарегистрированных медицинских изделий (за исключением медицинских изделий, связанных с источником энергии или оборудованных источником энергии) и (или) лекарственных препаратов, объединенных общей упаковкой, при условии сохранения вторичной (потребительской) упаковки или первичной упаковки лекарственного препарата в случае, если вторичная (потребительская) упаковка не предусмотрена, производителя (изготовителя) каждого из изделий и (или) лекарственных препаратов, входящих в указанные укладки, наборы, комплекты и аптечки, и при условии сохранения ее маркировки;
6) медицинские изделия, которые предназначены для диагностики заболеваний путем проведения исследований образцов биологического материала человека вне его организма, изготовлены в медицинской организации и применяются в медицинской организации, их изготовившей (далее — незарегистрированные медицинские изделия для диагностики in vitro).
(часть 5 в ред. Федерального закона от 30.04.2021 N 128-ФЗ)
5.1. Особенности обращения, включая особенности государственной регистрации, медицинских изделий, которые предназначены для применения в условиях военных действий, чрезвычайных ситуаций, предупреждения чрезвычайных ситуаций, профилактики и лечения заболеваний, представляющих опасность для окружающих, заболеваний и поражений, полученных в результате воздействия неблагоприятных химических, биологических, радиационных факторов, и которые разработаны в том числе по заданию федеральных органов исполнительной власти и федеральных государственных органов, в которых федеральным законом предусмотрена военная служба или приравненная к ней служба, а также медицинских изделий в случае их дефектуры или риска возникновения дефектуры в связи с введением в отношении Российской Федерации ограничительных мер экономического характера устанавливаются Правительством Российской Федерации.
(в ред. Федеральных законов от 01.04.2020 N 98-ФЗ, от 08.03.2022 N 46-ФЗ)
(часть 5.2 введена Федеральным законом от 30.04.2021 N 128-ФЗ)
6. Порядок ввоза на территорию Российской Федерации медицинских изделий в целях государственной регистрации устанавливается уполномоченным федеральным органом исполнительной власти. Порядок ввоза на территорию Российской Федерации медицинских изделий, указанных в пунктах 1, 2 и 5 части 5 настоящей статьи (за исключением медицинских изделий, указанных в подпунктах «а», «в» и «г» пункта 11 статьи 4 Соглашения о единых принципах и правилах обращения медицинских изделий (изделий медицинского назначения и медицинской техники) в рамках Евразийского экономического союза от 23 декабря 2014 года), устанавливается Правительством Российской Федерации.
(в ред. Федерального закона от 30.04.2021 N 128-ФЗ)
7. Ввоз на территорию Российской Федерации и вывоз с территории Российской Федерации медицинских изделий в рамках проведения допинг-контроля осуществляется в порядке, установленном Правительством Российской Федерации.
8. В целях государственной регистрации медицинских изделий в порядке, установленном уполномоченным федеральным органом исполнительной власти, проводятся оценка соответствия в форме технических испытаний, токсикологических исследований, клинических испытаний и экспертиза качества, эффективности и безопасности медицинских изделий, а также испытания в целях утверждения типа средств измерений (в отношении медицинских изделий, относящихся к средствам измерений в сфере государственного регулирования обеспечения единства измерений, перечень которых утверждается уполномоченным федеральным органом исполнительной власти). Действие данных требований может быть изменено в отношении участников экспериментального правового режима в сфере цифровых инноваций в соответствии с программой экспериментального правового режима в сфере цифровых инноваций, утверждаемой в соответствии с Федеральным законом от 31 июля 2020 года N 258-ФЗ «Об экспериментальных правовых режимах в сфере цифровых инноваций в Российской Федерации», с учетом требований, установленных правом Евразийского экономического союза.
(в ред. Федерального закона от 02.07.2021 N 331-ФЗ)
Производство медицинских изделий в ДНР, ЛНР, Запорожской и Херсонской областях до 01.01.2025, осуществляется без учета требований, установленных ч. 8.1 ст. 38 (ФЗ от 17.02.2023 N 16-ФЗ).
8.1. Производство медицинских изделий, подлежащих государственной регистрации, а также медицинских изделий, которые изготовлены по индивидуальным заказам пациентов, к которым предъявляются специальные требования по назначению медицинских работников, должно соответствовать требованиям к внедрению, поддержанию и оценке системы управления качеством медицинских изделий в зависимости от потенциального риска их применения, утвержденным Правительством Российской Федерации. Порядок организации и проведения инспектирования производства медицинских изделий на соответствие указанным требованиям устанавливается Правительством Российской Федерации. Методика определения размера платы за проведение такого инспектирования утверждается уполномоченным федеральным органом исполнительной власти.
(часть 8.1 введена Федеральным законом от 30.04.2021 N 128-ФЗ)
9. За совершение уполномоченным федеральным органом исполнительной власти действий, связанных с осуществлением государственной регистрации медицинских изделий и регистрации медицинских изделий в соответствии с международными договорами и актами, составляющими право Евразийского экономического союза, взимается государственная пошлина в соответствии с законодательством Российской Федерации о налогах и сборах.
(часть 9 в ред. Федерального закона от 30.04.2021 N 128-ФЗ)
10. В порядке, установленном Правительством Российской Федерации, уполномоченный им федеральный орган исполнительной власти осуществляет ведение государственного реестра медицинских изделий и организаций (индивидуальных предпринимателей), осуществляющих производство и изготовление медицинских изделий, и размещает его на своем официальном сайте в сети «Интернет».
(в ред. Федерального закона от 25.11.2013 N 317-ФЗ)
11. В государственный реестр медицинских изделий и организаций (индивидуальных предпринимателей), осуществляющих производство и изготовление медицинских изделий, вносятся следующие сведения:
1) наименование медицинского изделия;
2) дата государственной регистрации медицинского изделия и его регистрационный номер, срок действия регистрационного удостоверения;
3) назначение медицинского изделия, установленное производителем;
4) вид медицинского изделия;
5) класс потенциального риска применения медицинского изделия;
6) код Общероссийского классификатора продукции по видам экономической деятельности;
(п. 6 в ред. Федерального закона от 30.04.2021 N 128-ФЗ)
7) наименование и место нахождения юридического лица — уполномоченного представителя производителя (изготовителя) медицинского изделия или фамилия, имя и (если имеется) отчество, место жительства индивидуального предпринимателя — уполномоченного представителя производителя (изготовителя) медицинского изделия;
(п. 7 в ред. Федерального закона от 30.04.2021 N 128-ФЗ)
8) наименование и место нахождения организации — производителя (изготовителя) медицинского изделия или фамилия, имя и (если имеется) отчество, место жительства индивидуального предпринимателя — производителя (изготовителя) медицинского изделия;
(п. 8 в ред. Федерального закона от 25.11.2013 N 317-ФЗ)
9) адрес места производства или изготовления медицинского изделия;
10) сведения о взаимозаменяемых медицинских изделиях;
11) иные сведения, определяемые Правительством Российской Федерации.
(п. 11 введен Федеральным законом от 30.04.2021 N 128-ФЗ)
11.1. На территории Российской Федерации допускается изготовление, хранение, применение, утилизация или уничтожение в порядке, установленном уполномоченным федеральным органом исполнительной власти, незарегистрированных медицинских изделий для диагностики in vitro при наличии у медицинской организации разрешения на применение такого медицинского изделия, предоставленного уполномоченным федеральным органом исполнительной власти, осуществляющим функции по контролю и надзору в сфере охраны здоровья. Порядок предоставления, переоформления, подтверждения и отмены разрешения на применение незарегистрированного медицинского изделия для диагностики in vitro, а также требования к медицинским организациям, в которых изготавливаются и применяются незарегистрированные медицинские изделия для диагностики in vitro, и требования к таким медицинским изделиям утверждаются Правительством Российской Федерации. За предоставление, переоформление и подтверждение разрешения на применение незарегистрированного медицинского изделия для диагностики in vitro, а также за проведение экспертизы качества, безопасности и эффективности незарегистрированного медицинского изделия для диагностики in vitro в целях предоставления разрешения на применение незарегистрированного медицинского изделия для диагностики in vitro или подтверждения указанного разрешения взимается государственная пошлина в соответствии с законодательством Российской Федерации о налогах и сборах.
(часть 11.1 введена Федеральным законом от 30.04.2021 N 128-ФЗ)
12. Фальсифицированное медицинское изделие — медицинское изделие, сопровождаемое ложной информацией о его характеристиках и (или) производителе (изготовителе).
(часть 12 введена Федеральным законом от 31.12.2014 N 532-ФЗ)
13. Недоброкачественное медицинское изделие — медицинское изделие, которое не соответствует требованиям безопасности и эффективности медицинских изделий, требованиям к их маркировке, нормативной, технической и эксплуатационной документации и которое не может быть безопасно использовано по назначению, установленному производителем (изготовителем).
(часть 13 в ред. Федерального закона от 30.04.2021 N 128-ФЗ)
14. Контрафактное медицинское изделие — медицинское изделие, находящееся в обороте с нарушением гражданского законодательства.
(часть 14 введена Федеральным законом от 31.12.2014 N 532-ФЗ)
15. Запрещается производство:
1) незарегистрированных медицинских изделий, за исключением медицинских изделий, указанных в части 5 настоящей статьи;
(п. 1 в ред. Федерального закона от 30.04.2021 N 128-ФЗ)
2) фальсифицированных медицинских изделий;
3) медицинских изделий, содержащих этиловый спирт, по месту осуществления производства фармацевтической субстанции спирта этилового (этанола) и (или) по месту осуществления производства этилового спирта;
(п. 3 введен Федеральным законом от 27.12.2019 N 481-ФЗ)
4) медицинских изделий, содержащих этиловый спирт, на основном технологическом оборудовании для производства этилового спирта, указанном в пункте 1.1 статьи 14.1 Федерального закона от 22 ноября 1995 года N 171-ФЗ «О государственном регулировании производства и оборота этилового спирта, алкогольной и спиртосодержащей продукции и об ограничении потребления (распития) алкогольной продукции».
(п. 4 введен Федеральным законом от 27.12.2019 N 481-ФЗ)
(часть 15 введена Федеральным законом от 31.12.2014 N 532-ФЗ)
15.1. В случае необходимости использования этилового спирта, в том числе фармацевтической субстанции спирта этилового (этанола), при производстве медицинских изделий, содержащих этиловый спирт, в качестве действующего и (или) вспомогательного вещества, а также в иных технологических целях должна быть использована только фармацевтическая субстанция спирта этилового (этанол).
(часть 15.1 введена Федеральным законом от 27.12.2019 N 481-ФЗ)
16. Запрещается ввоз на территорию Российской Федерации фальсифицированных медицинских изделий, недоброкачественных медицинских изделий и контрафактных медицинских изделий, а также незарегистрированных медицинских изделий, за исключением медицинских изделий, указанных в части 5 настоящей статьи.
(часть 16 введена Федеральным законом от 31.12.2014 N 532-ФЗ; в ред. Федерального закона от 30.04.2021 N 128-ФЗ)
17. Запрещается реализация фальсифицированных медицинских изделий, недоброкачественных медицинских изделий и контрафактных медицинских изделий, а также незарегистрированных медицинских изделий, за исключением медицинских изделий, указанных в части 5 настоящей статьи.
(часть 17 введена Федеральным законом от 31.12.2014 N 532-ФЗ; в ред. Федерального закона от 30.04.2021 N 128-ФЗ)
18. Фальсифицированные медицинские изделия и недоброкачественные медицинские изделия подлежат изъятию и последующему уничтожению или вывозу с территории Российской Федерации, а контрафактные медицинские изделия — изъятию и последующему уничтожению. Вывоз с территории Российской Федерации фальсифицированных медицинских изделий и недоброкачественных медицинских изделий осуществляется за счет лица, осуществившего их ввоз на территорию Российской Федерации.
(часть 18 введена Федеральным законом от 31.12.2014 N 532-ФЗ)
19. Фальсифицированные медицинские изделия и недоброкачественные медицинские изделия подлежат изъятию из обращения и уничтожению на основании решения владельца медицинских изделий, решения уполномоченного федерального органа исполнительной власти, осуществляющего функции по контролю и надзору в сфере охраны здоровья, или решения суда. Контрафактные медицинские изделия подлежат изъятию из обращения и уничтожению по решению суда. Порядок изъятия из обращения и уничтожения фальсифицированных медицинских изделий, недоброкачественных медицинских изделий и контрафактных медицинских изделий устанавливается Правительством Российской Федерации.
(часть 19 в ред. Федерального закона от 02.07.2021 N 314-ФЗ)
20. Расходы, связанные с изъятием из обращения и уничтожением фальсифицированных медицинских изделий, недоброкачественных медицинских изделий и контрафактных медицинских изделий, возмещаются их владельцем.
(часть 20 введена Федеральным законом от 31.12.2014 N 532-ФЗ; в ред. Федерального закона от 02.07.2021 N 314-ФЗ)
21. В условиях чрезвычайной ситуации и (или) при возникновении угрозы распространения заболевания, представляющего опасность для окружающих, а также в случае, если в течение тридцати календарных дней после принятия Правительством Российской Федерации решения о проведении мониторинга розничных цен на медицинские изделия на территориях субъектов Российской Федерации выявлен рост розничных цен на медицинские изделия на тридцать процентов и более, Правительство Российской Федерации вправе установить предельные отпускные цены производителей на медицинские изделия, предельные размеры оптовых надбавок и предельные размеры розничных надбавок к фактическим отпускным ценам производителей на медицинские изделия. Перечень медицинских изделий и порядок его формирования определяются Правительством Российской Федерации.
(часть 21 введена Федеральным законом от 26.03.2020 N 67-ФЗ)
22. В течение девяноста календарных дней со дня утверждения Правительством Российской Федерации перечня медицинских изделий, указанного в части 21 настоящей статьи, не допускаются реализация и отпуск включенных в него медицинских изделий по ценам, превышающим предельные отпускные цены производителей на указанные медицинские изделия, предельные размеры оптовых надбавок и предельные размеры розничных надбавок к фактическим отпускным ценам производителей с учетом налога на добавленную стоимость.
(часть 22 введена Федеральным законом от 26.03.2020 N 67-ФЗ)
23. В условиях чрезвычайной ситуации и (или) при возникновении угрозы распространения заболевания, представляющего опасность для окружающих, Правительство Российской Федерации вправе принять решение об установлении ограничений на осуществление оптовой и розничной торговли медицинскими изделиями, включенными в определяемый Правительством Российской Федерации перечень, на срок, не превышающий девяноста календарных дней со дня принятия указанного решения.
(часть 23 введена Федеральным законом от 01.04.2020 N 98-ФЗ)
24. В условиях введения в отношении Российской Федерации ограничительных мер экономического характера Правительство Российской Федерации вправе принять решение об установлении ограничений на вывоз из Российской Федерации медицинских изделий, ранее ввезенных в Российскую Федерацию с территорий иностранных государств, принявших решение о введении в отношении Российской Федерации ограничительных мер экономического характера.
(часть 24 введена Федеральным законом от 08.03.2022 N 46-ФЗ)
25. Производители медицинских изделий или организации, осуществляющие ввоз на территорию Российской Федерации медицинских изделий, не менее чем за шесть месяцев до планируемых приостановления или прекращения производства медицинских изделий или их ввоза на территорию Российской Федерации уведомляют об этом федеральный орган исполнительной власти, осуществляющий функции по контролю и надзору в сфере здравоохранения.
(часть 25 введена Федеральным законом от 26.03.2022 N 64-ФЗ)
Перед выпуском в обращение медицинских изделий необходимым требованием является оформление декларации о соответствии ГОСТ Р. Наличие этого документа позволяет маркировать продукцию единым знаком качества.
Декларация соответствия на медицинские изделия – что это?
Это официальный документ, который подтверждает соответствие продукции законодательным актам и требованиям безопасности и качества. Действует ДС только на территории РФ. Перечень изделий, для которых обязательно оформление ДС, определен Постановлением №982.
Список включает в себя:
- Мед технику;
- Приборы диагностические и терапевтические;
- Медицинские объекты, изготовленные из полимеров;
- Имплантаты;
- Контрольно-измерительные приборы;
- Материалы для хирургии;
- Стоматологические продукты;
- Инструменты медицинского назначения;
- Протезно-ортопедические товары.
Нужна ли декларация соответствия на медицинские изделия в 2021 году?
С января 2021 года вступили в силу изменения (Приказ №478 ) в отношении декларирования мед изделий. Новые правила незначительно отличаются от прежних. Декларация соответствия все так же необходима для медицинских товаров, требования к заявителю, правила отбора образцов для испытаний, сама процедура оформления ДС не изменились. Сейчас сертифицирующие органы не могут самостоятельно зарегистрировать ДС в реестре, это сделает Росаккредитация.
Для кого обязательна декларация соответствия на мед. изделия?
Оформление ДС по ГОСТу обязательно для следующих товаров:
- Мед инструменты;
- Медицинская техника;
- Хирургические приспособления;
- Имплантаты;
- Линзы, корректирующие зрение;
- Приборы для наркоза;
- Товары для ухода за больными;
- Протезно-ортопедические товары;
- Зондирующие инструменты;
- Измерительная аппаратура;
- Стоматологические изделия и материалы;
- Приборы для терапевтической и диагностической деятельности;
- Ветеринарные приборы;
- Стеклянная тара медицинского назначения;
- Изделия из полимеров.
Полный перечень подлежащей декларированию продукции указан в Постановлении №982.
Какие медицинские изделия не подлежат государственной регистрации с последующим декларированием?
Не подлежат проверке в виде получения ДС следующие медицинские товары:
- Изготовленные по индивидуальным заказам пациентов;
- Санитарно-гигиеническая продукция для ухода за детьми;
- Санитарно-гигиенические изделия из металла, резины или пластмасс;
- Не перечисленные в Постановлении №982.
Если продукция не входит в списки подлежащих обязательному декларированию по системе ГОСТ, то производитель имеет право на оформление добровольного сертификата ГОСТ Р.
Где оформить декларацию ГОСТ Р на медицинские изделия?
Оформлять ДС можно только в специализированных центрах, имеющих аккредитацию на данный вид услуг. Соответствие нормам безопасности проверяется проведением испытаний, в протоколе фиксируются показатели и затем соотносятся значения желаемых и фактических данных. Перед получением декларации важно оформить регистрационное удостоверение на продукцию.
Как получить декларацию соответствия на медизделие?
Чтобы оформить ДС на медизделие, нужно пройти следующие процедуры:
- Сбор необходимых документов;
- Отбор образцов для испытаний;
- Проведение тестовых испытаний;
- Сбор комплекта доказательных документов и материалов;
- Передача заявления и регистрационного досье в сертифицирующий центр;
- Выезд на производство (при необходимости проверяется производственная линия и условия хранения;
- Принятие решения о выдаче ДС;
- Выдача готового документа.
Особенности оформления ДС по ГОСТ Р
Медицинская декларация выдается и действует только на территории России. Срок действия документа составляет 3 года или до окончания реализации партии. ДС оформляется как на отечественные изделия, так и на иностранные, импортные товары без данной декларации запрещено ввозить на территорию РФ. Сертификаты соответствия и ДС имеют одинаковую юридическую силу и обязательно регистрируются в едином реестре.
Какой список документов необходим для получения декларации соответствия ГОСТ Р на медицинские изделия?
Процедура оформления ДС ГОСТ возможна при наличии документов:
- Заполненная по образцу декларация;
- Заявление установленной формы;
- Учредительные и регистрационные данные производителя и заявителя (продавца, импортера);
- Сертификаты (ИСО, ГОСТ);
- Декларации и сертификаты соответствия;
- Регистрационное удостоверение;
- Протоколы проведенных испытаний;
- Подтверждение безопасности продукции;
- Технические условия;
- Другие, при необходимости.
Полный пакет необходимых материалов зависит от разновидности объекта, области применения и материалов.
Качество медицинского оборудования напрямую влияет на здоровье пользователей. В РФ сертификация является обязательной процедурой, без которой выпуск в обращение запрещен. Существует перечень объектов, подлежащих декларированию по ГОСТ.
Для чего нужен сертификат соответствия на медицинское оборудование?
Сертификатом соответствия (СС) производитель подтверждает безопасность техники. Использование оборудования без СС запрещено законом, работодателю за это грозит штраф или приостановка деятельности компании.
Существует разделение на 3 класса по опасности техники, для каждого класса предусмотрены свои показатели безопасности. Цель проверок и документа – подтверждение выполнения показателей безопасности по данному классу:
- 1 – умеренный риск;
- 2а – средняя степень риска;
- 2б – повышенный риск для людей;
- 3 – высокий риск.
Какая продукция должна быть сертифицирована?
Перечень техники состоит из:
- Травматологического оснащения;
- Измерительные приборы;
- Замещающие органы и системы организма;
- Для стоматологии и зубопротезирования;
- Осветительные приборы;
- Для стерилизации и дезинфекции;
- Устройств функциональной диагностики;
- Младенческих инкубаторов;
- Электрических рефлекторов;
- Томографов;
- Приборов для врачебных кабинетов и палат;
- Рентгеновской техники;
- Др.
Процедура сертификации медицинского оборудования в России
Процедура получения СС состоит из этапов:
- Выбор организации, проводящей услугу;
- Отбор образцов оборудования для испытаний;
- Проведение тестовых испытаний для образцов;
- Составление протокола;
- Формирование пакета документов;
- Принятие решения о выдаче разрешительного документа;
- Выдача СС и занесение сведений в единый реестр.
Список необходимых испытаний зависит от вида техники и класса опасности.
Какие документы необходимы для сертификации медицинской техники?
Для процедуры сертификации необходимо подготовить следующие материалы:
- Учредительные и регистрационные документы (заверенные копии);
- Описание устройств;
- Технические документы;
- Паспорт оборудования;
- Протоколы проведенных испытаний;
- Руководство по эксплуатации;
- Подтверждение права собственности на производственную площадь (в собственности или аренде);
- Имеющиеся сертификаты, регистрационные удостоверения;
- Контракты на поставку (копии);
- Сопроводительные документы;
- Др.
Срок действия сертификата на медицинское оборудование
Сроком действия документа может быть период 1 или 3 года (в зависимости от схемы оценки). На партию товара срок не устанавливается, он действует до конца реализации указанной партии. В это время могут проводиться инспекционные проверки организации, оцениваться производственный процесс. Цель – проверка, соблюдаются ли изготовителем требования государственных стандартов. По окончанию действия документа заявитель может подать заявку на продление. СС действует на территории России.
Как получить сертификат для медицинского оборудования?
Получить документ могут российские и иностранные изготовители, процедура происходит через органы, имеющие аккредитацию по ГОСТ. Срок оформления СС зависит от вида оборудования и от объема необходимых тестовых испытаний. Испытания могут проводиться только в аккредитованных лабораториях, в большинстве случаев свои лаборатории есть в сертифицирующих органах. Помимо сертификата, изготовители обязаны зарегистрировать медицинские изделия в Росздравнадзоре, без регистрационного удостоверения запрещена реализация и использование техники в РФ. Если техника подпадает под действие ТР ТС 020, то дополнительно необходимо оформить декларацию или сертификат по техрегламенту.
Как проверить сертификат на медицинское оборудование?
В реестре Росаккредитации можно в любой момент проверить статус СС на медоборудование. В документе указаны регистрационный номер, организация, оформившая СС, и ее контактные данные. Любой пользователь или заинтересованное лицо может обратиться с запросом в данную организацию о получении информации о сертификате, изготовителе и выпущенном оборудовании.
Сертификат на медицинские изделия и лекарственные средства – это официальный документ, который свидетельствует о соответствии объекта требованиям безопасности. Целью проверки является защита потребителей от некачественных товаров.
Сертификация медицинских изделий – что это?
Лекарственные средства, как и другие медицинские товары, способны повлиять на жизнь и здоровье людей. Поэтому Росздравнадзор тщательно контролирует данные изделия и предусматривает несколько форм их проверки.
Сертификация – обязательная проверка всех медицинских товаров, которые реализуются на территории РФ, произведены они могут быть в любой стране.
Сертификат соответствия (СС) оформляется на бланке установленного образца, содержит следующую информацию:
- Название лекарственного средства или изделия;
- Сведения о заявителе и производителе;
- Нормативы, по которым изготовлен товар;
- Доказательные материалы, подтверждающие безопасность;
- Сведения сертификационного органа;
- Номер документа и дата выдачи.
Медицинские изделия, подлежащие обязательной сертификации
Практически вся медицинская продукция подлежит либо обязательной сертификации, либо добровольному декларированию. Перечни изделий и порядок проведения определены Постановлением Правительства №982 и Правилами №36. Нужно оформлять СС на изделия:
- Лекарственные средства, выпускаемые на территории РФ;
- Ввозимые на территорию РФ.
Отсутствие документа является административным нарушением, влечет штрафы и изъятие продукции.
Какие медицинские изделия подлежат добровольной сертификации?
Лекарственные изделия, на которые можно оформить добровольный СС:
- Средства без индивидуальной упаковки, созданные не для прямой реализации;
- Субстанции для производства лекарств;
- Вакцины, сыворотки, иммунобиологические средства.
Добровольный и обязательный документы идентичны, одинаковы порядки их оформления и регистрации. В самом бланке СС есть указание, обязательна ли форма оформления.
Для чего проводят регистрацию и сертификацию изделий медицинского назначения?
Основной целью оформления разрешительного документа является выполнения требований законодательства страны. Его наличие позволит легко проходить проверки, которые обязательны для медицинских товаров. Отсутствие СС чревато штрафами и изъятием партии. Среди других причин:
- Повышение лояльности со стороны потребителей;
- Формирование положительного имиджа среди компаний-конкурентов;
- Укрепление своих позиций;
- Возможность подачи заявки на тендерах федерального масштаба;
- Увеличение рынков сбыта;
- Увеличение продаж.
Порядок сертификации лекарственных средств в России
Лекарственные средства имеют стандартный порядок сертификации, он включает в себя:
- Выбор сертифицирующего органа – это может быть сторонняя организация с соответствующей аккредитацией;
- Выбор схемы оценки;
- Отбор образцов для тестирования;
- Идентификация изделий;
- Испытания в аккредитованной лаборатории с составлением протокола полученных показателей;
- Оценка производства (если предусмотрено схемой);
- Анализ полученных сведений и материалов, принятие решения;
- Выдача сертификата или мотивированного отказа;
- Инспекционные контроль (если предусмотрено).
Предусмотрено несколько схем сертификации:
- 7с – на партию;
- 3 и 3а – на серийное производство.
Срок действия документа ограничен – до 3-х лет, после чего требуется повторная процедура прохождения оценки изделий.
От чего зависит стоимость сертификации ЛС и мед изделий?
Стоимость процедуры рассчитывается индивидуально. Факторы, от которых зависит цена:
- Вид продукции;
- Класс опасности товаров;
- Размер предприятия;
- Схема сертификации;
- Наличие документации.
Для консультации по поводу цены и срока оформления документа можно обратиться к экспертам сертифицирующего центра. Каждый центр предлагает комплексную помощь для получения СС.
Где получить сертификат соответствия на медицинские изделия?
Заниматься оформлением сертификатов по ГОСТу может не любая организация, а только имеющая соответствующую аккредитацию. Процедура подразумевает участие трех сторон: заявителя (чаще всего это производитель, но может быть и поставщик), сертифицирующего органа и испытательной лаборатории. Чтобы не ошибиться с выбором исполнителя, стоит обратить внимание на:
- Опыт работы организации;
- Спектр медицинских товаров, с которыми проводятся процедуры;
- Наличие собственной испытательной лаборатории;
- Гибкое ценообразование;
- Разумные сроки выполнения оценки (слишком быстрые сроки могут быть сомнительны).
Какие документы необходимы для получения сертификата на медицинские изделия?
Перечень необходимых документов состоит из:
- Заявления установленного образца;
- Регистрационное удостоверение;
- Копию учредительных и организационных документов;
- Копия устава;
- Протоколы проведенных испытаний;
- Образцы изделий;
- Технические условия;
- Разрешение на применение товара в лечебной практике;
- Доверенность от компании-изготовителя на предоставление своих интересов на территории РФ (для импортных товаров);
- Подтверждающие безопасность изделия материалы;
- Другие по запросу.
Все копии должны быть заверенными, пакет документов сшит.
Перед началом продажи медицинских изделий необходимо провести оценку их соответствия актуальным требованиям законодательства. Разрешительные документы гарантируют высокое качество продукции, ведь от этого зависит жизнь и здоровье пациентов.
Сертификация медицинских изделий — процедура проверки, которую осуществляют уполномоченные органы (центры) по заявлению производителей или импортеров товаров.
Оборудование, подлежащее обязательной сертификации
Изделия, применяемые в медицине, принято классифицировать в зависимости от величины потенциальной опасности:
- 1 класс (пониженная степень риска) — микроскопы, медицинские весы;
- 2а класс (средняя степень риска) — лабораторная аппаратура;
- 2б класс (повышенная степень риска) — дефибрилляторы, кардио анализаторы;
- 3 класс (высокая степень риска) — эндопротезы.
Обязательной проверке качества и безопасности подлежат следующие изделия:
- медицинская техника;
- хирургические имплантаты;
- терапевтические и диагностические приборы;
- медицинские наборы;
- контрольно-измерительная аппаратура;
- материалы, применяемые в хирургии;
- медицинские инструменты;
- стоматологические материалы;
- протезно-ортопедические товары;
- медицинские изделия из полимеров.
Какой документ требуется получить?
Предприятиям, решившим заняться изготовлением и реализацией медицинской продукции, требуется оформить разрешительную документацию. Такое же правило действует и в отношении компаний, организующих импортные поставки на территорию России.
В перечень обязательных документов могут входить:
- декларация ГОСТ Р (по требованиям ПП РФ №982) — разрабатывается в ходе оценки соответствия диагностических перчаток, масок, инструментов, бумажных изделий, перевязочных средств, мебели, стоматологических материалов, трубок, катетеров, линз;
- регистрационное удостоверение Росздравнадзора (по правилам ПП РФ №1416) — оформляется на все медицинские товары (в т.ч. лекарственные средства) и является бессрочным;
- свидетельство об утверждении типов средств измерений (СУТСИ) — необходимо получать на измерительные приборы (тонометры, кардиографы, весы).
Декларация ГОСТ Р
Документ имеет юридическую силу в России в течение трех лет с момента регистрации. Перед выдачей декларации присваивается уникальный номер, который вносится в единый реестр ФСА. Это основной показатель подлинности разрешения и его легитимности.
Оценка соответствия проводится в специализированных лабораториях и представляет собой проверку качества и безопасности товара, а также анализ регистрационных и технических сведений предпринимателя.
Декларация составляется на простом белом листе формата А4, содержит важные данные о продукции и ее изготовителе (заявителе).
Обратиться в уполномоченный центр для регистрации документа могут только компании-резиденты РФ, зарубежным предприятиям нужен официальный представитель из нашей страны.
Схемы декларирования
Контроль безопасности продукции проводится по одной из установленных схем:
- проверка серийного выпуска — схемы 1Д, 2Д, 3Д, 4Д;
- экспертиза партии — 5Д;
- оценка единицы продукции — 6Д.
Административная ответственность
Отсутствие разрешительной документации на продукцию является серьезным правонарушением, которое ведет к административной ответственности. При нанесении вреда человеку возможно и уголовное наказание для недобросовестного предпринимателя.
Чаще всего к нарушителям применяются:
- штрафы крупных размеров;
- изъятие контрафакта;
- временное прекращение работы на срок до 3-х месяцев.
Для чего нужна добровольная оценка качества?
Добровольный сертификат — выступает важным конкурентным преимуществом для ведения бизнеса. Он свидетельствует о соответствии изделий требованиям национальных стандартов, которые предприниматель может выбрать самостоятельно.
Например, это могут быть:
- ГОСТ ISO 13485-2017 Изделия медицинские. Системы менеджмента качества. Требования для целей регулирования;
- ГОСТ 31508-2012 Изделия медицинские. Классификация в зависимости от потенциального риска применения. Общие требования.
Добровольная оценка позволит выигрывать государственные тендеры и осуществлять крупные поставки в медицинские учреждения, в которых предъявляются строгие требования к продукции.
Этапы оформления сертификата
Алгоритм контрольной процедуры состоит из следующих шагов:
- обращение в надежный центр, который выдает только легитимную документацию;
- заполнение заявки на оказание услуги и передача необходимой документации;
- идентификация продукции и выбор подходящей схемы;
- подписание договора между сторонами и оплата аванса;
- изучение предоставленных данных экспертами центра;
- отбор образцов для проведения испытаний и осуществление экспертизы;
- подготовка протоколов;
- регистрация готового документа в реестре ФСА и отправка клиенту (проводится при условии, что исследования подтвердили соответствие изделий действующим нормативам).
Далее на товар наносится маркировка знаком соответствия “РСТ”. Теперь предприниматель может осуществлять оборот продукции по всей России на законных основаниях.
Сбор сведений
Предпринимателю необходимо подготовить регистрационные и производственные сведения для проведения процедуры:
- копии свидетельств налоговых органов — ИНН, ОГРН;
- стандарты изготовления (ГОСТ, ТУ, СТО);
- название, свойства и назначение продукции;
- руководство по эксплуатации, технический паспорт;
- сертификат на систему управления качеством (если есть);
- контракт на ввоз импорта (если есть).
Услуги по сертификации медицинских изделий
Центр “ГОСТ Центр” готов помочь предпринимателям в быстром получении необходимой документации. В нашем коллективе работают квалифицированные профессионалы, которые хорошо знакомы с особенностями оценки соответствия продукции в регионе.
Изделия, подлежащие обязательной регистрации
Согласно статье 28 323-ФЗ госрегистрация изделий медицинского назначения обязательна для всех видов товаров, которые используются в целях:
- профилактики заболеваний и патологий;
- диагностики;
- лечения;
- реабилитации;
- предотвращения или прерывания беременности;
- других медицинских целях.
Классы потенциального риска
В зависимости от сложности, области применения и других факторов медицинские изделия различаются по критерию класса риска. Он представляет собой степень потенциального вреда, который товар способен нанести здоровью пациента даже при условии соблюдения рекомендаций производителя относительно его использования. По этому признаку выделяются классы:
- 1 – низкий;
- 2а – средний;
- 2б – повышенный;
- 3 – высокий.
Товары, не подлежащие постановке на учет
Невзирая на то, что действующее законодательство устанавливает требование об обязательной регистрации почти для всех товаров медицинского назначения, из этого правила есть некоторые исключения. Согласно пункту 5 статьи 38 323-ФЗ госрегистрация медицинского изделия не потребуется, если речь идет о товаре для использования конкретным пациентом. Их изготавливают в соответствии с характером его заболевания и рекомендациями его лечащего врача. Это минимизирует возможность применения такого продукта другими пациентами, поэтому ставить его на учет не нужно.
Порядок процедуры
С 1 января 2022 года Российская Федерация, как и другие страны-участницы Евразийского экономического союза, полностью перешла на общесоюзные правила госрегистрации и сертификации медицинских товаров. Они регулируются положениями решения Коллегии ЕЭК от 12.02.2016 № 46. В соответствии с ним регистрация медизделий в Росздравнадзоре предполагает последовательную реализацию следующих шагов.
- Заявитель организует необходимые испытания, оформляет и собирает требуемые документы и составляет регистрационное досье.
- Заявитель выбирает референтное государство, где будет подаваться заявки на госрегистрацию медицинского изделия, и государства признания, где медицинское изделие сможет обращаться после получения регистрационного удостоверения.
- Заявитель подает регистрационное заявление и досье в Росздравнадзор.
- Эксперты ведомства анализируют степень полноты и корректности оформления пакета и при отсутствии ошибок назначают экспертизу товара.
- Сотрудники уполномоченной экспертной организации проводят экспертизу, в рамках которого изучаются данные, содержащиеся в регистрационном досье. Результаты экспертизы оформляются экспертным заключением, которое передается в Росздравнадзор.
- Специалисты аккредитованной организации выполняют инспекцию производства медицинского изделия, в рамках которой проверяют соответствие оборудования, технологических процессов и других параметров установленным нормативам.
- Аккредитованная медицинская организация проводит клинические испытания, в ходе которых исследуются качество, безопасность и действенность продукта. Результаты испытаний оформляются протоколами установленного образца, которые передаются в Росздравнадзор.
- Сотрудники Росздравнадзора изучают полный пакет документов по изделию и, в случае положительной рекомендации, выданной медицинскими экспертами, размещают его в межгосударственной информационной системе для ознакомления сотрудниками уполномоченных органов государств признания.
- Сотрудники уполномоченных органов государств признания изучают документацию по изделию и принимают решение о ее согласовании.
- Потом Росздравнадзор принимает решение по государственной регистрации продукта. Если оно окажется положительным, сотрудники ведомства оформляют и выдают заявителю регистрационное удостоверение и вносят информацию об изделии в Единый реестр зарегистрированных продуктов.
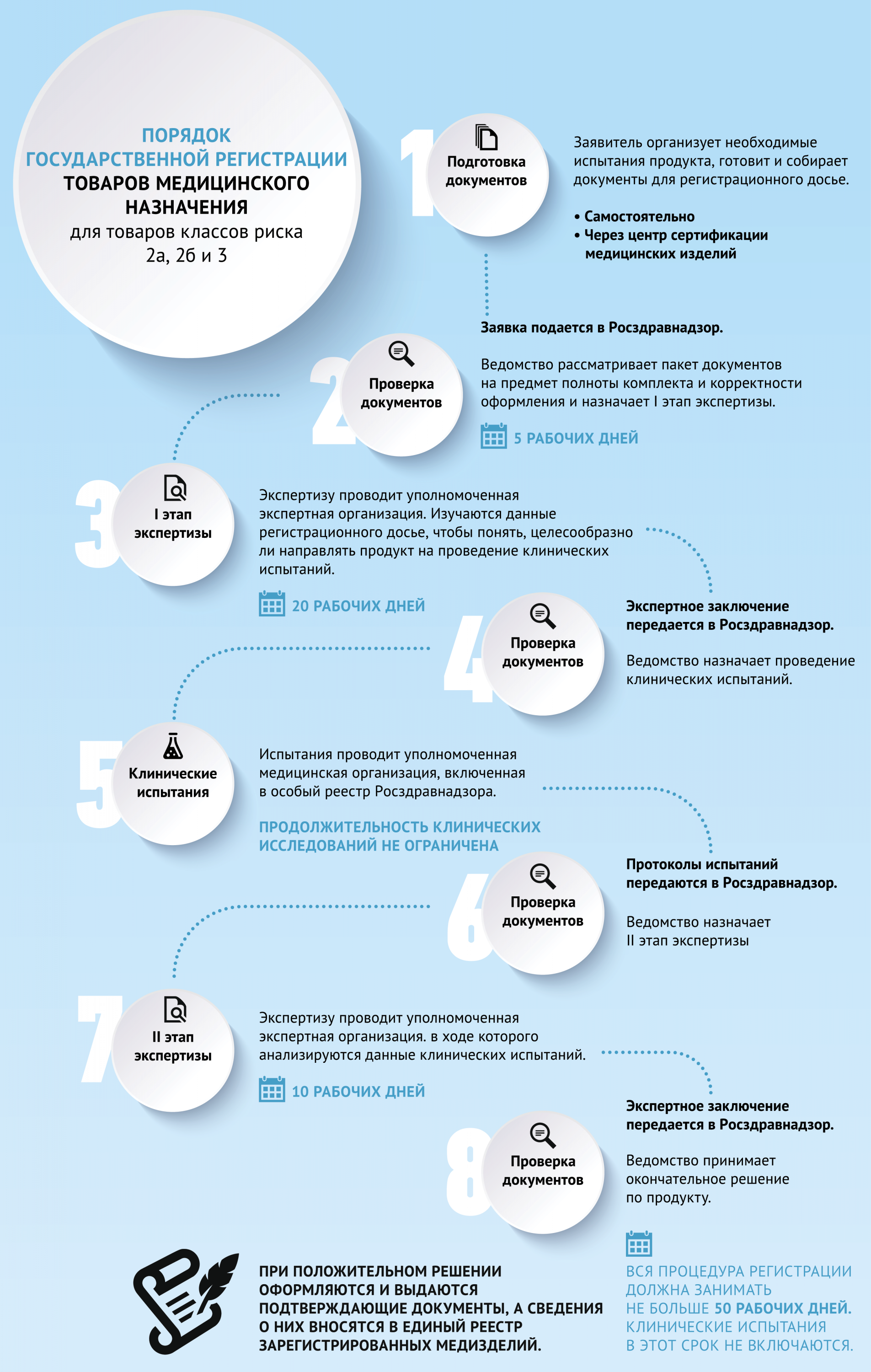
Сроки
Максимальная длительность регистрации складывается из нескольких компонентов:
- изучение комплекта документации на изделие и решение о начале процедуры регистрации – 5 рабочих дней;
- экспертиза регистрационного досье – 60 рабочих дней;
- инспекция производства изделия – 90 рабочих дней;
- согласование экспертного заключения уполномоченными органами государств признания – 30 календарных дней;
- принятие решения о регистрации изделия – 10 рабочих дней;
- оформление регистрационного удостоверения -10 рабочих дней.
Размер государственной пошлины
Согласно статье 333.32.2 действующего Налогового кодекса РФ стоимость регистрации становится суммой двух государственных пошлин:
- за выдачу регистрационного удостоверения – 11 тысяч рублей;
- за выполнение экспертизы – от 72 до 184 тысяч рублей в зависимости от класса риска изделия.
Распространенные ошибки при самостоятельной подаче заявки
Процесс взаимодействия с контролирующими государственными органами часто не приносит нужного результата. Основными ошибками, которые допускают заявители при самостоятельном обращении в Росздравнадзор, становятся:
- неполный или неправильно оформленный пакет документов на изделие;
- непроведение всех нужных испытаний;
- нарушение сроков подачи заявки;
- непредоставление дополнительной документации по запросу ведомства;
- другие ошибки.
Преимущества работы с нашим центром
Уполномоченный центр «Безопасность» предлагает помощь в регистрации врачебного изделия любой категории. Наши эксперты много лет сотрудничают с Росздравнадзором в этой области и знают все требования, действующие на текущий момент. Благодаря этому Ваша заявка будет удовлетворена с первого раза, и Вы оперативно пройдете государственную регистрацию, выведете товар на рынок и займете достойное место среди ведущих производителей.
Отправить заявку на услугу
Изделия и оборудование медицинского предназначения в связи с повышенным риском для здоровья человека относятся к категории важных объектов технического регулирования. Это определяет сложность сертификации в этой сфере производства и реализации: контролирующие органы уделяют повышенное внимание товарам для медицинских целей — ведь от этого зависит безопасность человеческой жизни.
Разрешительные документы для медицинских товаров в Российской Федерации
Применение продукции специального назначения для использования в медицинских целях в РФ возможно только после обязательного регистрационного учета. Этот процесс осуществляется путем оформления документации в службе по надзору в Минздравсоцразвития РФ, а также получения сертификата соответствия на конкретный вид товара.
Также в случае необходимости оформляется отказное письмо системы ГОСТ Р, которое свидетельствует о том, что по закону обязательная сертификация медицинской техники данного типа не нужна.
Сертификация изделий для медицинской деятельности
Подтверждение соответствия медоборудования стандартам качества – обязательный этап для доказательства соответствующего качества продукции данной категории по нормативам системы ГОСТ Р.